Biologie Verirrte Proteine
Wenn es in Proteinen zu genetischen Veränderungen kommt, kann das schwere gesundheitliche Folgen haben. Auch Veränderungen in Abschnitten der Moleküle, die als funktionslos galten, können Ursache für Erkrankungen sein. Dafür gibt es nun eine Erklärung
Als Macie ein Jahr alt war, bemerkten ihre Eltern zum ersten Mal dieses Zucken in ihren Ärmchen und Beinchen. Wenn sie hungrig oder müde war, schien das immer öfter vorzukommen. Als Macie dann eines Tages für einige Momente ganz benommen war, nahm auch der Kinderarzt die Sorgen ernst: Macie litt unter Epilepsie.
Es folgten fast zehn Jahre mit wechselnden Diagnosen und Therapien, bis Macie kurz vor ihrem elften Geburtstag erfuhr, dass sie diese epileptischen Anfälle hat, weil ihr Gehirn nicht genügend mit Energie versorgt wird. Sie leidet am sogenannten GLUT1Defizit-Syndrom. Der wichtigste Energielieferant für unser Gehirn ist Zucker. Nur kann dieser nicht ohne Hilfe die geheimnisvolle Barriere überwinden, die Blutbahn und Gehirngewebe trennt. Diese Blut-Hirn-Schranke lässt nur bestimmte Substanzen in unser empfindliches Denkorgan.
Macies Eltern erfuhren nun, dass das Protein, das die Zuckermoleküle aus dem Blut durch die Schranke ins Gehirn transportiert, bei ihrer Tochter anders zusammengebaut ist als bei den meisten Menschen. Das Merkwürdige jedoch war, dass bei ihr nur ein einziger Baustein, an einer scheinbar unwichtigen Stelle des Proteins, ausgetauscht war.
Auch wenn Macies GLUT1-Defizit-Syndrom äußerst selten ist, ist die Veränderung in ihren Genen eine von Zehntausenden, bei denen einzelne Proteine, die elementaren Bausteine und Funktionsträger unseres Körpers, genetisch bedingt verändert sind. Und das kann zu schweren Krankheiten führen. Uns machte besonders stutzig, dass diese Veränderungen auch an Stellen der Proteine fatal sein können, denen bislang nicht viel Bedeutung zugemessen wurde und die als mehr oder weniger funktionslos galten. Doch wie können bestimmte Veränderungen in diesen Regionen zu Krankheiten führen?

Proteine verrichten ihre Aufgaben oft nicht ganz alleine, sondern in Teamwork. Dabei können sie sich zu stabilen Komplexen zusammenschließen, in anderen Fällen geht es auch nur um die kurze Weitergabe von Informationen. Es gibt Beispiele, bei denen genau die scheinbar funktionslosen Regionen der Proteine für diesen kurzen Informationsaustausch verantwortlich sind.
Und so wollten wir herausfinden, ob sich diese Interaktionen bei normalen und bei mutierten Proteinen voneinander unterscheiden.
Da Proteine mit Hunderten anderen Proteinen wechselwirken können und oft nicht bekannt ist, welches mit welchem interagiert, ist ein spezielles Analyseverfahren notwendig. Bestens geeignet dafür: das Massenspektrometer. Selbst aus der komplexesten Proteinmischung kann damit die Masse jedes einzelnen bestimmt und daraus die Zusammensetzung ermittelt werden.
Tatsächlich zeigte sich, dass viele der mutierten Proteine ihre Präferenzen verändert hatten und nun mit anderen Proteinen interagierten. Unter diesen mutierten Formen waren drei besonders auffällig, da sie mit den gleichen Proteinpartnern neue Kontakte eingingen. Bei allen mutierten Proteinen handelte es sich um sogenannte Transmembranproteine. Diese haben ihren Platz in der Membran, die jede Zelle wie eine schützende Burgmauer umgibt. Transmembranproteine kann man sich wie Burgwächter vorstellen, die entscheiden, welche Substanzen in die Zelle hineindürfen und welche nicht oder auch welche Informationen in die Zelle weitergegeben werden sollen.
Eine weitere Gemeinsamkeit: In allen drei Fällen waren die gleichen Proteinbausteine gegeneinander ausgetauscht worden, nämlich Prolin gegen Leucin. Außerdem lagen als Folge in den mutierten Proteinen stets zwei Leucinmoleküle nebeneinander. Und dies ist bemerkenswert, denn zwei benachbarte Leucine sind für spezielle Transportproteine ein Signal. Es ist eine Anweisung, die Transmembranproteine von der Zellmembran ins Zellinnere zu leiten.
Sie sehen gerade einen Platzhalterinhalt von YouTube. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenEigentlich ist dieser Prozess, die sogenannte Endozytose, völlig normal und lebenswichtig. Doch kann er fatale Folgen haben, wenn er fehlerhaft ausgelöst wird. In diesem Fall ist das Signal falsch – die Transmembranproteine senden es nur deshalb aus, weil es in einem ihrer eigentlich für unbedeutend gehaltenen Abschnitte zu einer Mutation gekommen ist. Die Folge: Die Transportproteine bekommen die falsche Anweisung und ziehen die Burgwächter von ihren Posten ab.
Könnte das auch der Grund für den fehlerhaften Zuckertransport in Macies Kopf sein? Ist Macies Variante des Zuckertransporters vielleicht gar nicht funktionsunfähig, sondern sitzt einfach nur an der falschen Stelle?
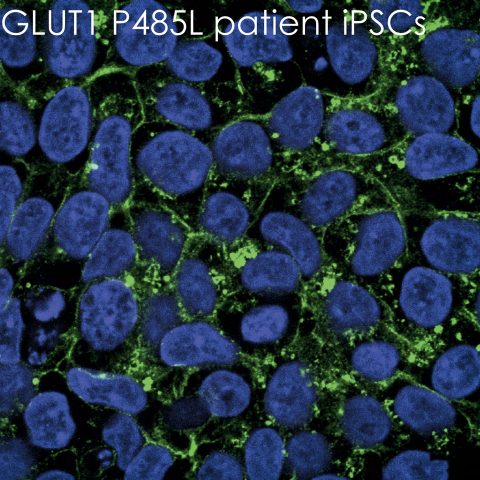
Um das zu untersuchen, kultivierten wir Zellen aus der Haut von Macies Oberarm – und markierten ihre Glukosetransporter mit fluoreszentem Farbstoff. Unter dem Mikroskop sehen die aneinander gedrängten Zellen mit gesunden Transmembranproteinen, jede mit dem fluoreszierenden Protein in der Burgmauer, fast aus wie Honigwaben (siehe Beitragsbild). Doch Macies Zellen sind gänzlich mit einem Punktmuster durchsetzt. Die Burgmauer ist fast nicht zu erkennen – und das heißt, dass sich ihre Burgwächter tatsächlich nicht an ihren Posten befinden. Der Zuckertransport ist unmöglich.
Unsere Vermutung war also richtig. „Aber sofort drängte sich die nächste Frage auf”, sagt Matthias Selbach, Leiter der Arbeitsgruppe Proteom Dynamik am Berliner Max-Delbrück-Centrum für Molekulare Medizin. „Nun wollten wir wissen, ob Macies Transmembranprotein wieder Zucker transportieren kann, wenn wir es in die Zellmembran zurückbringen.”
Dazu entfernten wir zunächst mithilfe einer molekularbiologischen Methode jene Transportproteine, die das Transmembranprotein fälschlicherweise ins Zellinnere gelenkt hatten. Und tatsächlich: Die Burgwächter kehrten auf ihre Position in der Zellwand zurück! Wenn man diese Zellen nun mit Zucker fütterte, nahmen sie auch deutlich mehr davon auf, als jene Zellen, in denen wir die Transportproteine nicht entfernt hatten. Die Erklärung für Macies Leiden war also gefunden: Der Transportweg für Zucker in ihr Gehirn war unterbrochen, weil die entsprechenden Transmembranproteine nicht dort waren, wo sie gebraucht wurden – in der Zellmembran.
Sind vielleicht noch viel mehr Menschen krank, nur weil eigentlich funktionsfähige Proteine an die falsche Stelle transportiert werden? Tatsächlich stießen wir beim Durchforsten von Datenbanken auf elf Proteinvarianten, in denen Transmembranproteine das Signal aussenden, von ihrem Posten abgezogen zu werden.
Macie bekämpft ihre Symptome, indem sie ihr Gehirn mit einem anderen Energieträger versorgt. Sie verzichtet fast vollständig auf Zucker und Kohlenhydrate und bringt so ihren Körper dazu, stattdessen Fett als neue Energiequelle zu erschließen, die sogenannten Ketonkörper. Dadurch lindert sie die Auswirkungen ihres Leidens, das eigentliche Problem ist damit aber natürlich nicht behoben. Menschen, die unter anderen Folgen solch fehlerhaft lokalisierter Proteine leiden, haben diese Möglichkeiten oft nicht. Doch kann dieses neue Wissen über fehlgeleitete Proteine eine wichtige Grundlage sein, zukünftig auch andere Erkrankungen mithilfe zielgerichteter Medikamente zu behandeln.
Waisen der Medizin
Es gibt Krankheiten, die sind so selten, dass sie kaum erforscht sind. Doch das ändert sich
Jenes GLUT1-Defizit-Syndrom, unter dem Macie leidet, wurde 1991 entdeckt und ist extrem selten. Das ist typisch für die sogenannten seltenen Erkrankungen, die so heißen, wenn sie unter 2000 Menschen höchstens einmal auftreten.
Doch bedeutet dies nicht, dass nur wenige davon betroffen sind. Denn es gibt rund 8000 seltene Erkrankungen. So schätzen Experten, dass allein in Deutschland rund vier Millionen Menschen damit leben müssen. 75 Prozent davon sind Kinder, fast ein Drittel stirbt vor dem fünften Lebensjahr. Etwa 80 Prozent der Leiden sind genetisch bedingt.
Für die Betroffenen bedeutet das stets ein mehrfaches Schicksal. So ist die Diagnose seltener Erkrankungen viel langwieriger – im Schnitt besteht erst nach fünf bis sechs Jahren Gewissheit. Doch selbst dann können Patienten nicht immer auf Hilfe hoffen. Denn wegen der geringen Fallzahlen sind diese Krankheitsbilder entsprechend wenig erforscht. Weil Pharmakonzerne überdies die Kosten für die Entwicklung von Medikamenten scheuen, schließt sich hier ein Teufelskreis. Menschen mit solchen Erkrankungen werden deshalb auch „Waisen der Medizin“ genannt: Sie haben niemanden, der ihnen hilft.
Doch es tut sich was. So lassen neue Forschungen vermuten, dass es sich bei Massenleiden wie Rheuma, Diabetes oder auch Parkinson in Wahrheit um eine Vielzahl seltener Erkrankungen handelt, deren gezielte Behandlung vielversprechend ist und am Ende viel mehr Menschen hilft. Überdies profitieren Unternehmen von Förderprogrammen der Europäischen Union und investieren zunehmend in diese Marktlücke. Mit Erfolg: Von allen Medikamenten mit neuem Wirkstoff, die in Deutschland 2014 bis 2018 auf den Markt kamen, gehörte bereits ein Drittel zu den „Waisenmedikamenten“ (Orphan Drugs).
Von Joachim Schüring
